XXYY is a rare X and Y chromosome variant which is not greatly understood.
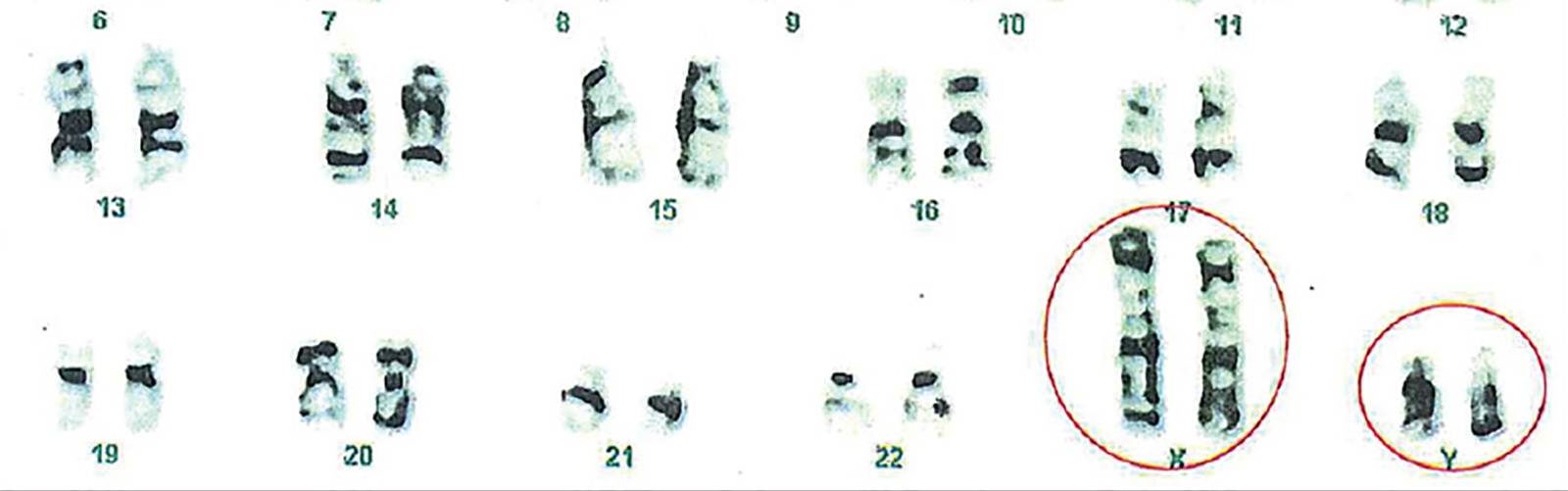
XXYY syndrome is a sex chromosome anomaly in which males have an extra X and Y chromosome. Human cells usually contain two sex chromosomes, one from the mother and one from the father. Usually, females have two X chromosomes and males have one X and one Y chromosome. The appearance of at least one Y chromosome with a properly functioning SRY gene makes a male. Therefore, humans with XXYY are genotypically male. Males with XXYY syndrome have 48 chromosomes instead of the typical 46. This is why XXYY syndrome is sometimes written as 48,XXYY syndrome or 48,XXYY.
While XXYY Syndrome is similar to other conditions such as Klinefelter’s Syndrome (boys with an extra X chromosome) and XYY Syndrome, boys and men with XXYY Syndrome are more significantly impacted in their development, behaviour and medical issues.
Who is affected
XXYY syndrome is estimated to affect 1 in 18,000 males.
Causes
This condition is not inherited; it usually occurs as a random event during the formation of reproductive cells (eggs and sperm). An error in cell division called nondisjunction results in a reproductive cell with an abnormal number of chromosomes. In XXYY syndrome, the extra sex chromosomes almost always come from a sperm cell. Nondisjunction may cause a sperm cell to gain two extra sex chromosomes, resulting in a sperm cell with three sex chromosomes (one X and two Y chromosomes). If that sperm cell fertilizes a normal egg cell with one X chromosome, the resulting child will have two X chromosomes and two Y chromosomes in each of the body’s cells.
In a small percentage of cases, XXYY syndrome results from nondisjunction of the sex chromosomes in a 46,XY embryo very soon after fertilization has occurred. This means that a normal sperm cell with one Y chromosome fertilized a normal egg cell with one X chromosome, but right after fertilization nondisjunction of the sex chromosomes caused the embryo to gain two extra sex chromosomes, resulting in a XXYY syndrome embryo.
The cause of XXYY syndrome is still not completely understood. Since extensive studies have not been conducted on the cause of XXYY syndrome, there is currently very little information about whether or not there are environmental, hereditary or other factors which can result in this chromosome pattern. There is no evidence that parents of one XXYY child are more likely to have other children with sex chromosomal variations.
Signs and symptoms
XXYY syndrome is often mistaken for other syndromes. The most common symptoms and characteristics that would be noticeable by parents, teachers, medical professionals and other treatment providers are:
- Developmental delays
- Speech impairment or delay
- Tall, considering family history
- Behavior outbursts & mood swings
- Learning disabilities
- Intellectual impairment
- ADD or ADHD symptoms
- Autism, autism spectrum, PDD-NOS
- Scoliosis
- Clinodactyly (Curved-in pinky fingers)
(Not all boys and men with XXYY Syndrome experience all of these symptoms)
The majority of children with XXYY syndrome have some developmental delays and learning disabilities. Most males with XXYY syndrome have an IQ that ranges from 70-80, although this is greatly variable, they often have some degree of difficulty with speech and language development. Learning challenges, especially those that are language-based, are very common in males with this disorder. Affected males seem to perform better at tasks focused on math, visual-spatial skills such as puzzles, and memorization of locations or directions. Some boys with XXYY syndrome have delayed development of motor skills such as sitting, standing, and walking that can lead to poor coordination. Affected males have higher than average rates of behavioural disorders, such as attention deficit hyperactivity disorder (ADHD); mood disorders, including anxiety and bipolar disorder; and autism spectrum disorder, which affects communication and social interaction.
Extra copies of genes on the X chromosome interfere with male sexual development. Adolescent and adult males with this condition typically have small testes that do not produce enough testosterone, which is the hormone that directs male sexual development. A shortage of testosterone during puberty can lead to reduced facial and body hair, poor muscle development, low energy levels, and an increased risk for breast enlargement (gynecomastia). Because their testes do not function normally, males with XXYY syndrome have an inability to father children (infertility).
XXYY syndrome can affect other parts of the body as well. Males with XXYY syndrome are often taller than other males their age with an average adult height of 6 feet 4 inches (193 cm). They tend to develop a tremor that typically starts in adolescence and increases with age. Dental problems are frequently seen with this condition; they include delayed appearance of the primary (baby) or secondary (adult) teeth, thin tooth enamel, crowded and/or misaligned teeth, and multiple cavities. As affected males get older, they may develop a narrowing of the blood vessels in the legs, called peripheral vascular disease. Peripheral vascular disease can cause skin ulcers to form. Affected males are also at risk for developing a type of clot called a deep vein thrombosis (DVT) that occurs in the deep veins of the legs. Additionally, males with XXYY syndrome may have flat feet (pes planus), elbow abnormalities, abnormal fusion of certain bones in the forearm (radioulnar synostosis), allergies, asthma, type 2 diabetes, seizures, and congenital heart defects.
Many genes are found only on the X or Y chromosome, but genes in areas known as the pseudo autosomal regions are present on both sex chromosomes. Extra copies of genes from the pseudo autosomal regions of the extra X and Y chromosome contribute to the signs and symptoms of XXYY syndrome; however, the specific genes have not been identified.
Diagnosis
Prenatal testing is significantly increasing due to its availability and continued advancements. Many expectant parents are completely unaware that these tests can also identify X & Y sex chromosome variations. These tests can be obtained via analysis of the mother’s blood known as the Harmony test or NIPT. The results can give a percentage of likelihood of an XXYY diagnoses. Diagnoses isn’t confirmed until further testing is obtained. The options available are, the analysis of amniotic fluid via amniocentesis or analysis of tissue samples from a portion of the placenta via chorionic villus sampling (CVS). These tests are invasive and can have a small risk of causing a miscarriage. Expectant parents have the option to wait until the baby is born and confirm a diagnosis through the baby’s blood sample.
Diagnosing XXYY syndrome postnatally requires a genetic test called a karyotype. The test is done by drawing blood and an analysis is done on the cells of the blood to determine the boy or man’s chromosomal make-up. A karyotype is the only way to know for certain that a boy or man has XXYY syndrome if a prenatal diagnosis isn’t confirmed.
In fact, unless diagnosed in utero, or in early childhood, many boys and men are not diagnosed with XXYY syndrome until their late teens, when low testosterone symptoms begin to manifest. This is largely due to other diagnoses given throughout childhood which have been used to rationalize their behaviors (i.e. autism, ADD), medical issues (i.e. low muscle tone, heart problems) and academic results (i.e. learning disabilities, ADD).
Treatment
Expectant parents with an XXYY prenatal diagnoses should experience a normal pregnancy with no additional concerns or complications because of the diagnoses.
A team approach to therapy, treatment, early intervention and schooling is recommended for all individuals with XXYY syndrome. Not all boys and men with XXYY syndrome will show and be affected with all the signs and symptoms but it is important that regular health checks are done to rule out and treat any potential health concerns. Early intervention can make a significant positive impact on a child’s life.
Medical evaluation at the time of a diagnosis depends on the age of the individual but should include a complete medical history and examination with an emphasis on features requiring monitoring and intervention. In infants and children, cardiac evaluation for congenital heart defects and mitral valve prolapse, renal ultrasound, ophthalmologic evaluation, and orthopaedic evaluation for flat feet, scoliosis, and elbow dysplasia are recommended. Aggressive preventative dental care and hygiene should also be implemented. Brain MRI, EEG, and sleep study should be strongly considered depending on clinical symptoms. Adolescents and adults should also be screened for other associated medical problems including Type 2 diabetes, hypothyroidism, osteoporosis, and obesity. A high index of suspicion for deep vein thrombosis, pulmonary embolus, and non-Hodgkin lymphoma should be maintained for various presenting symptoms.
Endocrinology consultation should be obtained upon diagnosis to inform parents and individuals about options and timing of androgen treatments and updated practices in the treatment of hypogonadism and infertility. If hypogonadism is present, testosterone treatment should be considered in all individuals regardless of cognitive abilities due to positive effects on bone health, muscle strength, fatigue, and endurance, with possible mental health/behavioural benefits as well. For those with significant mental health problems such as bipolar or psychotic disorders, testosterone treatment should be initiated slowly with close observation by the mental health team and avoidance of concurrent changes to other psychotropic medications or environmental settings. With any medical treatment individual choice and a collaborative and holistic approach is very important before deciding on a treatment plan.
Since almost all children with XXYY have developmental delays and learning difficulties, a comprehensive neurodevelopmental evaluation is warranted at the time of diagnosis, including assessments by psychology (cognitive and social–emotional development), speech/language therapy, occupational therapy, and physical therapy. Consultation with a developmental paediatrician, psychiatrist, or neurologist to develop a treatment plan including therapies, behavioural interventions, educational supports, and psychotropic medications for behavioural and psychiatric symptoms should be arranged and initiated if required. Common diagnoses such as learning disability/ID, ADHD, autism spectrum disorders, mood disorders, tic disorders, and other mental health problems should be considered, screened for, and treated. Good responses to standard medication treatments for inattention, impulsivity, anxiety, and mood instability are seen in this group and such treatment can positively impact academic progress, emotional wellbeing and long-term outcome.
Poor fine motor coordination and the development of intention tremor can make handwriting slow and laborious, and occupational therapy and keyboarding should be introduced at an early age to facilitate schoolwork and self-help skills. Educational difficulties should be evaluated with a full psychological evaluation to identify discrepancies between verbal and performance skills and to identify individual academic needs. Expressive language skills are often affected throughout the lifespan and speech therapy interventions targeting expressive language skills, dyspraxia, and language pragmatics may be needed into adulthood. Adaptive skills (life skills) are a significant area of weakness necessitating community-based supports for almost all individuals in adulthood.
Working in a collaborative and holistic manner is vital and has huge benefits to everyone.
With proactive management and effective treatment the impacts XXYY has on a child and individuals life can be greatly decreased.
